Dataset Information
| Overview | |
|---|---|
| Title | Molecular characterization of selectively vulnerable neurons in Alzheimer’s Disease (human) |
| Sub dataset | This study has two datasets. This is the EC brain region dataset. |
| Design | Single-nucleus RNA-sequencing of post-mortem brain tissue from donors spanning the range of AD progression. |
| Abstract | Alzheimer’s disease (AD) is characterized by the selective vulnerability of specific neuronal populations, the molecular signatures of which are largely unknown. To identify and characterize selectively vulnerable neuronal populations, we used single-nucleus RNA sequencing to profile the caudal entorhinal cortex and the superior frontal gyrus – brain regions where neurofibrillary inclusions and neuronal loss occur early and late in AD, respectively – from individuals spanning the neuropathological progression of AD. We identified RORB as a marker of selectively vulnerable excitatory neurons in the entorhinal cortex, and subsequently validated their depletion and selective susceptibility to neurofibrillary inclusions during disease progression using quantitative neuropathological methods. We also discovered an astrocyte subpopulation, likely representing reactive astrocytes, characterized by decreased expression of genes involved in homeostatic functions. Our characterization of selectively vulnerable neurons in AD paves the way for future mechanistic studies of selective vulnerability and potential therapeutic strategies for enhancing neuronal resilience. The brain tissue used in this study were sourced from the Biobank for Aging Studies (LIM-22) in the Department of Pathology at the University of Sao Paulo, as well as from the Neurodegenerative Diseases Brain Bank in the Memory and Aging Center at the University of California, San Francisco. Detailed neuropathological, clinical and genetic data are available under request. Please contact [email protected] and [email protected]. |
| Year | 2020 |
| Reference | Molecular characterization of selectively vulnerable neurons in Alzheimer's disease Nat Neurosci. 2021;24(2):276-287. |
| PubMed | 33432193 |
| Source |
Expression data source: GSE147528_EC Metadata source: GEO sample description |
| Sample description | |
| Organism | Homo sapiens |
| Type | snRNA-seq |
| Samples | |
| Count | |
| Cell types | |
| Metadata availability | |
Sample Metadata
Dimensional Reduction Plot Colored by [ Cell Type ]
Dimensional Reduction Plot Colored by Expression of [ ]
Dimensional Reduction Plot Colored by Sample Metadata [ ]
COLOR BY
SAMPLE SELECTOR
PLOT CONTROL
OVERVIEW

Differential Expression Analysis
Select StrategyComparisonCell type to show comparison results.
Select Cutoff to check for result availabilities of PPI network, Enrichment Analysis, and Drug Screen.
log2FC = FDR =
| Number of DEGs by cutoffs | |log2FC| > | |||
|---|---|---|---|---|
| 0.25 | 0.1 | 0 | ||
| 0.05 | ||||
| FDR < | 0.10 | |||
| 0.20 | ||||
| Gene (log2FC ≥ 0) | log2FC | FDR |
|---|---|---|
| SELECT A COMPARISON TO VIEW THE UP-EXPRESSED GENES | ||
| Gene (log2FC < 0) | log2FC | FDR |
|---|---|---|
| SELECT A COMPARISON TO VIEW THE DOWN-EXPRESSED GENES | ||
Enrichment Analysis
| KEGG Pathways Human | Overlap | P | FDR |
|---|---|---|---|
| Gene Ontology - Biological Process | Overlap | P | FDR |
|---|---|---|---|
Drug Screening
| Significantly inversely related | ES | FDR |
|---|---|---|
| Significantly positively related | ES | FDR |
|---|---|---|
| Signature ID | |
|
|---|---|---|
| Perturbagen ID | ||
| Name | ||
| SMILES | ||
| INCHI KEY | ||
| DrugBank ID | ||
| Cell line | ||
| Dose | ||
| Time | ||
| # Replicates | ||
| QC Pass | ||
| High Quality | ||
| Tool |
Cell-cell Interaction Analysis
Click a cell-cell interaction from the heatmap to view the over-represented ligand-receptor pairs.
Significant LRIs in +
| Ligand | Receptor | Mean | P |
|---|---|---|---|
| SELECT A CELL-CELL INTERACTION FROM THE HEATMAP TO VIEW THE OVER-REPRESENTED LIGAND-RECEPTOR PAIRS | |||
All LRIs in descending order by # of significant means
| Ligand | Receptor | # CCI | CCI net |
|---|---|---|---|
| SELECT AN ANALYSIS TO VIEW ALL THE OVER-REPRESENTED LIGAND-RECEPTOR PAIRS | |||
Protein-Protein Interaction Network for DEGs in the Selected DE Comparison Drug-Target Network for Selected Drug Perturbation Network for Selected Compound Ligand-Receptor Interaction Network for Selected Cell-Cell Interaction Cell-Cell Interaction Network for Selected Ligand-Receptor Interaction
PLOT CONTROL
LEGEND
Sorry, we cannot retrieve data from the server at this time. Please refresh the page later. If the issue persists, please contact us.
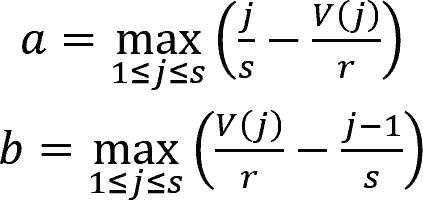 where j = 1, 2, ..., s were the indexes of DEGs sorted by their ranks in the gene profiles of the perturbation in ascending order.
The rank of gene j is denoted by V(j), where 1 ≤ V(j) ≤ r, with r being the total number of genes in the profile.
Then, ESup/down was set to aup/down if aup/down > bup/down, and was set to -bup/down if bup/down > aup/down.
Permutation tests repeated 10000 times using randomly generated gene lists with the same n(up) and n(down) as the DEGs were performed to measure the one-tailed p values of the ES scores.
Depending on the sign of the ES, the one-tailed p value measures either the left-tail (ES < 0) or the right-tail (ES > 0). P values were FDR adjusted and FDR < 0.05 were considered significant.
where j = 1, 2, ..., s were the indexes of DEGs sorted by their ranks in the gene profiles of the perturbation in ascending order.
The rank of gene j is denoted by V(j), where 1 ≤ V(j) ≤ r, with r being the total number of genes in the profile.
Then, ESup/down was set to aup/down if aup/down > bup/down, and was set to -bup/down if bup/down > aup/down.
Permutation tests repeated 10000 times using randomly generated gene lists with the same n(up) and n(down) as the DEGs were performed to measure the one-tailed p values of the ES scores.
Depending on the sign of the ES, the one-tailed p value measures either the left-tail (ES < 0) or the right-tail (ES > 0). P values were FDR adjusted and FDR < 0.05 were considered significant.